
To get access to the DART-MS:
- Read this page carefully! You should read it over a few times and make sure you understand all aspects. If you have questions, find me or call me on Teams.
- Prepare a sample and find Ivan. No appointment is necessary, but you can send a Teams chat message or call/text 607 255 0709 to make sure I’m available before coming to the facility, if you want.
- I will ask you questions about the contents of this page. If your responses lead me to believe that you did not read this page carefully, you will not be able to complete your training and will have to try again at a later time. If you seem to know what you are talking about, I will walk you through running your sample at the instrument and you will be cleared to use the DART on your own.

What is DART?
The “DART” spectrometer is an Exactive Orbitrap mass spectrometer equipped with a Direct Analysis in Real Time (DART) ion source. The Eaxctive is a high-resolution, accurate-mass capable instrument, so you can use it to confirm or propose elemental compositions. DART is an open-to-atmosphere, surface-desorption ionization method that works for small to medium sized organic and organometallic analytes. It is very quick and requires little to no sample preparation. DART can also be used in the analysis of synthetic polymers and plastics. It will not work for very polar molecules, like sugars and peptides.
MS Basics
NMR is NMR. All NMR spectrometers operate on the same fundamental physical principles, and all analytes behave in predictably similar ways. Mass spectrometry (MS), on the other hand, is an umbrella term that describes a multitude of approaches to measuring a physical property (voltage, time, frequency, etc.) that is proportional to the mass-to-charge ratio of the analyte. The physics are different, the applications are different, and analytes do NOT behave the same way, sometimes in strange and unpredictable ways. Because of this, you need to carefully consider the chemical and physical properties of your analyte, as well as published precedents, before selecting a particular ion source and mass spectrometer for analysis. It is also insufficient to state that you “did mass spec” on a sample; you need to be specific in terms on ionization and detection to convey useful information.
Mass spec can be a useful tool for
- measuring molecular mass
- confirming the presence of elements with stable isotopes
- confirming or proposing elemental composition
- identifying structural subunits
- following reactions
- confirming or identifying isotopic labelling or kinetic isotope effects.
Mass spec should NOT be used to measure purity, except in special cases.
DART Basics
DART is an open-to-atmosphere, surface-desorption ionization method that works for most of small to medium sized organic and organometallic analytes. It is very quick and requires little to no sample preparation. DART can also be used in the analysis of synthetic polymers and plastics. It doesn’t work well for very apolar molecules, like unsaturated hydrocarbons. You should probably use our GC-Exactive instrument for those samples. DART does NOT work for very polar molecules, like sugars and peptides, but we can help you analyze those with ESI.

Inside the DART ion source, an inert gas (nitrogen or helium) passes over a heater, through a plasma, and, finally, an electrode that traps charged species. This generates neutral, excited, “metastable” gas atoms/molecules, which interact with the air in the sampling region to create a “magic soup” of excited gas and water vapor. The choice of DART gas and its temperature are user controllable parameters. A small diaphragm vacuum pump keeps the DART gas and neutral analytes moving through the ion source while charged analytes are pulled into the mass spectrometer orifice by electrostatic attraction.
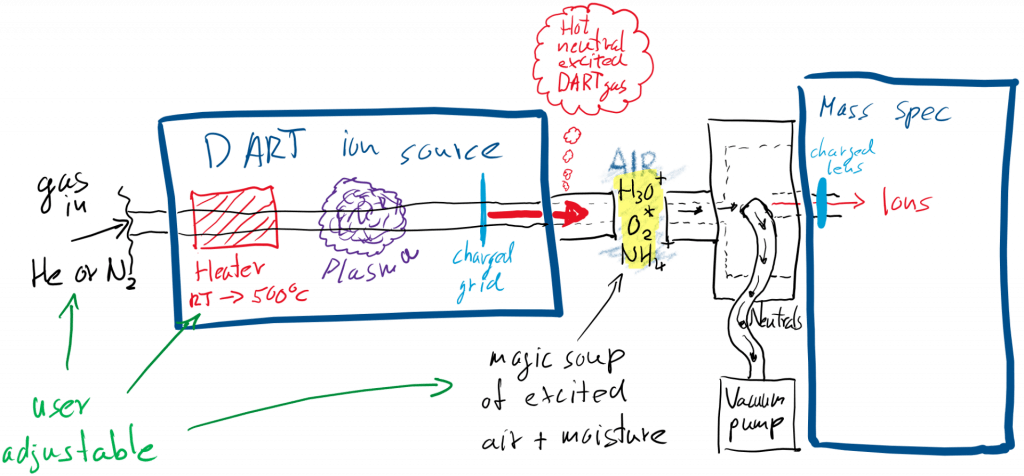
To analyze a sample, you simply place the analyte into the “magic soup” using a surface of your choice. We typically use the closed end of a melting point capillary, but anything that fits into the sampling gap can be analyzed. Ionization happens in two distinct steps: 1) the analyte needs to desorb from the surface and 2) it has to react with the “magic soup” to form an ion.

The desorption step limits the utility of DART to volatile and semi-volatile analytes with molecular weights up to ~1000 Da but the mass limit depends strongly on the polarity/volatility of the molecule. For example, we have detected polyaromatics in the 1500 Da range, while even small peptides will not work.
Positive Ion Mode
With ionic compounds the unmodified molecular cation [M+] is typically observed.
Neutral analytes typically ionize by adduct formation, and you can expect to see multiple adducts at the same time. Proton adducts [M+H]+ are by far the most common. In more concentrated samples [2M+H]+ dimers, or, for mixtures, [M1+M2+H]+ mixed dimers are possible. In coordinating solvents, like methanol and acetonitrile, we have seen [M+solventn+H]+ adducts. The second most common ionization mechanism is ammonium adduct [M+NH4]+ formation. While ammonium adducts can be dominant in some cases, they are usually observed secondary to proton adducts. For some analytes oxygen adducts with the formulae [M+H2O+H]+, [M+OH]+, [M+O]+ and [M+O2]+ are also possible. Changing the DART gas between helium and nitrogen may alter the identity and distribution of adducts. Penning ionization, which yields [M∙+] radical cations is also possible, especially for amines. Adducts with group I metal ions (Li+, Na+, K+) are NEVER observed.
While DART is a mild ionization method that typically yields molecular adduct ions, your analyte may decompose in the ion source to form fragment ions, especially at higher DART temperatures. These fragmentation processes do not follow the “classical” fragmentation mechanisms of the radical cations generated in electron impact (EI) ionization. They are the results of unimolecular, “acid-catalyzed,” gas-phase chemical reactions. Alcohols, ethers and esters may dehydroxylate/dealkoxylate to yield [M-OH]+ and [M-OR]+ carbocations, and alkyl halides (and presumably sulfonate esters) may show [M-X]+ ions. This is analogous to the first step in E1 and SN1 reactions, and the likelihood of fragmentation depends on the stability of the leaving group and resulting cation. Primary alcohols are unlikely to show fragmentation, while you may not be able to detect the molecular ion for benzoic esters. Other possible reactions include decarboxylation of beta-keto acids, N2 release from azides, and retro-Diels-Alder reactions. Organometallic coordination complexes can ionize by losing an anionic ligand to yield [M-L]+ ions. This is especially common for halide complexes, where ligand loss is often the predominant ionization mechanism.
Negative Ion Mode
With ionic compounds the unmodified molecular anion [M–] is typically observed.
Negative ion mode works very well for neutral analytes with acidic hydrogens; with proton loss as the primary ionization mechanism giving [M-H]– ions. In chlorinated solvents [M+Cl]– adducts have been reported, but we haven’t observed them on our instrument. Negative ion mode is much less general than positive ion mode, which makes it very useful for finding trace amounts of ionizable compounds.
Synthetic Polymers
At lower DART gas temperatures, you can desorb low molecular-weight components from synthetic polymers. This can be useful for identifying additives like plasticizers and UV protectants. At high DART gas temperatures, polymers may pyrolyze to yield distributions of oligomers that can be used to determine repeating units. This can be useful for characterizing co-polymers or identifying unknown plastics. DART cannot be used to determine the molecular weight distribution of polymers. If you want to do that, ask us about MALDI or ESI.
Adjusting DART Conditions
To detect your analyte in DART, you will need to adjust the acquisition conditions. Some analytes work well with minimal effort and some require extensive tweaking. We have given up trying to predict how any particular sample will go. You have control over the following:
- DART gas (helium or nitrogen)
- Temperature of DART gas
- Exposure time
- Additives
- Surface/matrix
- Sample prep
Dart Gas
You can select nitrogen or helium as the DART carrier gas. The two gases can have very different ionization profiles, in unpredictable ways. Some analytes work great in one and don’t ionize at all in the other. Helium is more expensive than nitrogen, so please start with nitrogen, and if you don’t see the mass you are looking for switch to helium. You will want to switch back and forth as you increase the gas temperature.
Dart Gas Temperature
The temperature of the DART gas is the most important user-controllable variable for generating good data. If the temperature is too low, the analyte will not desorb from the surface and therefore not ionize. If the temperature is too high, you will miss your analyte because it will desorb and ionize instantaneously. You need to find the temperature where the analyte desorbs at a steady rate over tens of seconds. This means that a sample containing multiple analytes may require analysis at multiple temperatures. Our rule of thumb is a starting temperature of 100°C below the MW of your compound, or 200°C, whichever is lower. Lower polarity requires lower temperatures and higher polarity requires higher temperatures. Keep in mind that higher temperatures also increase the probability of the fragmentation described above.
Dart Gas Exposure Time
The longer you hold the analyte in the gas stream, the hotter the capillary gets. Some analytes require tens of seconds of exposure before they desorb. If you don’t see your peaks immediately, simply waiting longer can work wonders.
Additives
You can alter the ionization profile by changing the composition of the “magic soup.” For example, opening a bottle of aqueous ammonia solution can increase the prevalence of ammonium adducts. Introducing a coordinating solvent can generate [M+solventn+H]+ adducts. The inventor of DART told me that introducing chlorinated solvents, like dichloromethane or chloroform, can yield [M+Cl]– adducts in negative ion mode, but we have never managed to get it to work. If you do, please let us know.
Surface
For the vast majority of samples, the closed end of a glass capillary works well as a sampling device. If you are having difficulties you can try using a metal spatula (BYO), metal mesh (ask us), wood or glass wool. Always collect a “blank” from your matrix before introducing your analyte.
Sample Prep
Solution-Phase Samples (preferred)
Solutions of your samples should be similar in concentration to NMR samples, although, unlike ESI, the exact concentration is unimportant. Unlike ESI, GC-MS, and MALDI type techniques, high-boiling solvents such as DMF and DMSO do not pose a problem since they are not introduced directly into the source. Prior user experience indicates that for some analytes the choice of the solvent has a noticeable effect on ionization efficiency. You will be dipping a glass capillary tube into your sample solution, so you only need a small amount (μL’s) but make as much as is easy for you to handle in case you need to repeat the analysis.
NMR Samples
NMR samples are perfect starting points for DART analysis. We recommend that you get a DART-MS on every NMR sample you run, but at the very least on every new compound.
Solid-State Samples
Use of solid samples (powders and crystals) is possible but discouraged based on prior experience. These samples can clog the inlet capillary and contaminate the spectrometer for extended periods. If dissolving the sample is not a viable option, or if solution samples did not yield acceptable results, samples can be introduced directly as solids but use as little solid as possible! Grind the sample finely with the closed end of the melting point capillary prior to analysis. Small pieces or “chunks” of sample should never be used because these can be pulled into the analyzer by the vacuum. Use very small amount of sample; the solid should be barely visible on the tip of the capillary. MS is a very sensitive technique, and in our experience, putting more solid on the end of the capillary will not improve your results. If you are having trouble with your sample, try changing the conditions instead of using more analyte.
Surface Desorption
Analytes may be desorbed directly from matrices or surfaces, such as glass- or metal-backed analytical TLC plates, silicon wafers, leaves, peels, paper, bugs etc. When planning a desorption experiment, keep in mind that the DART gas is hot (150 to 550 °C) and may melt, set on fire, or otherwise destroy a poorly chosen matrix/surface.
TLC Plates
DART can be used to get MS on analytical TLC spots using either of the following methods:
- Glass- or metal-backed TLC plates can be analyzed by holding them flat in the center of the DART source to let the DART gas skim the surface. For best results plates must be less than ¼ inch wide. Standard width (~¾”) TLC plates don’t work well for direct sampling.
- Scrape the spot into a vial and add solvent. Sample resulting solution as usual. It is not necessary to filter the sample. You can also scrape all of the silica off the plate in sections and use DART to analyze the resulting “fractions”.
Volatiles
Volatile analytes may be detected by simply bringing an object near the DART. In general, if you can smell it, you should be able to analyze it without direct contact with the DART plasma. This also means that volatiles from personal-care products, tobacco and other sources may interfere with analyses.
Air-Sensitive Analytes
Solutions should be in narrow vials under argon and sampled as usual. Alternatively, use a septum capped vial and a syringe (BYOS) and squeeze a very small drop of solvent to the tip of the syringe while holding it into the DART stream. For solid samples, a user reported good results from coating a melting point capillary with the sample and wrapping the tip of the capillary tightly with parafilm. Remove the parafilm just before inserting the capillary into the DART stream.
HOW TO DART
- If you see the login screen, log in with the credentials posted on the monitor bezel.
- The DART source is controlled from a Microsoft Edge window. If the window isn’t on the screen, start Microsoft Edge by double clicking the icon on the desktop of taskbar.
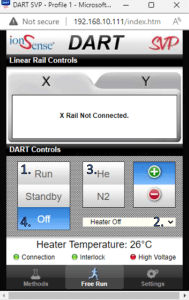
- Click the “Run” button (1. on image)
- Adjust the DART gas temperature from the drop down box (2. on image)
- If you can’t see the ions you are looking for, try changing the DART gas to helium (3. on image)
- Remember to turn off the DART source when you are done with your samples (4. on image)
- The mass spectrometer is controlled from the Exactive Tune software (if it isn’t running, start it by double clicking the iTunes-like music notes icon on the desktop or taskbar):
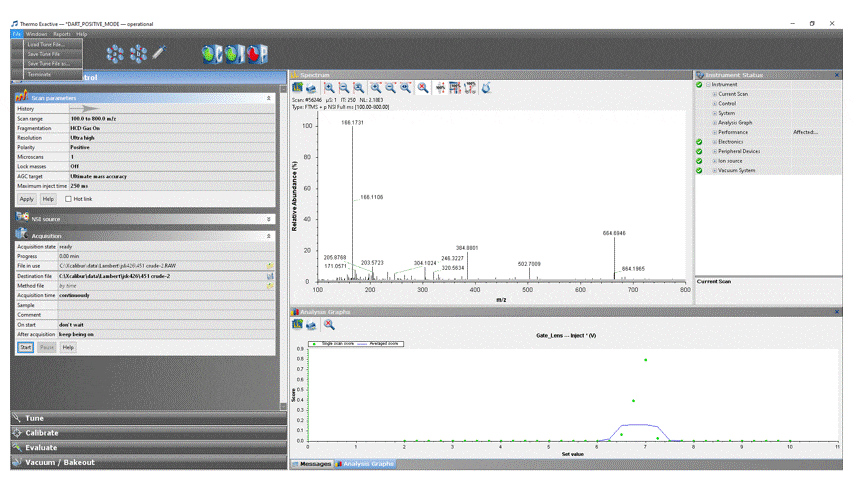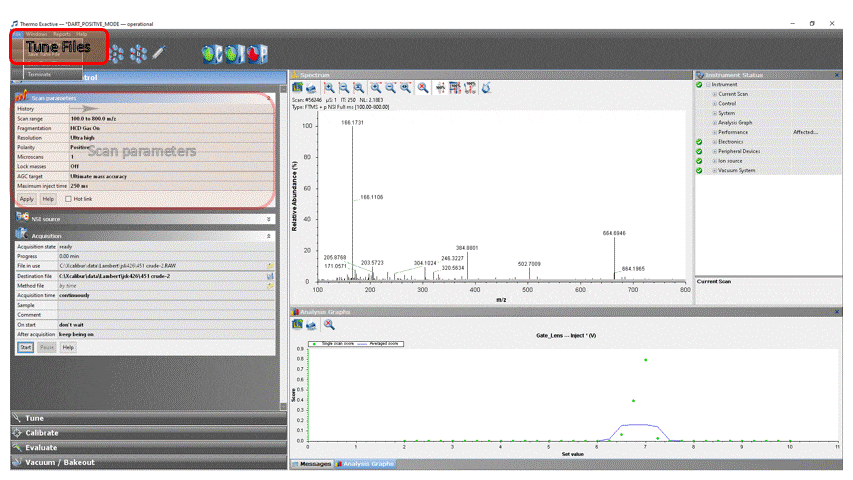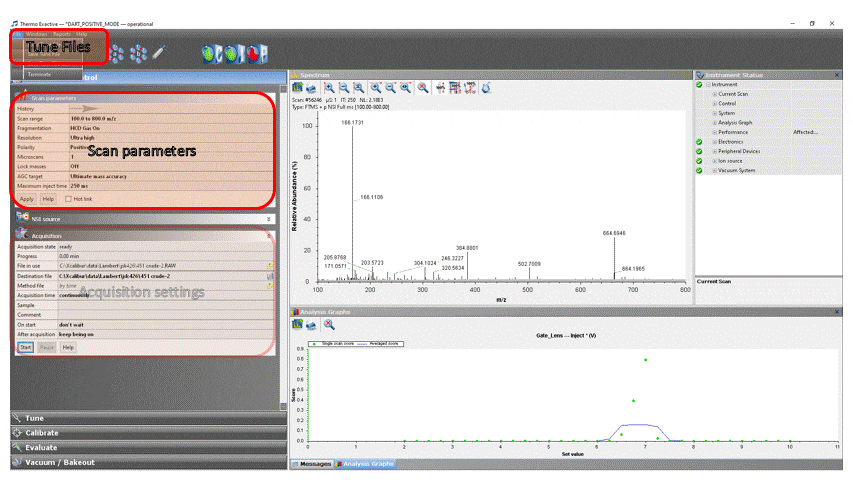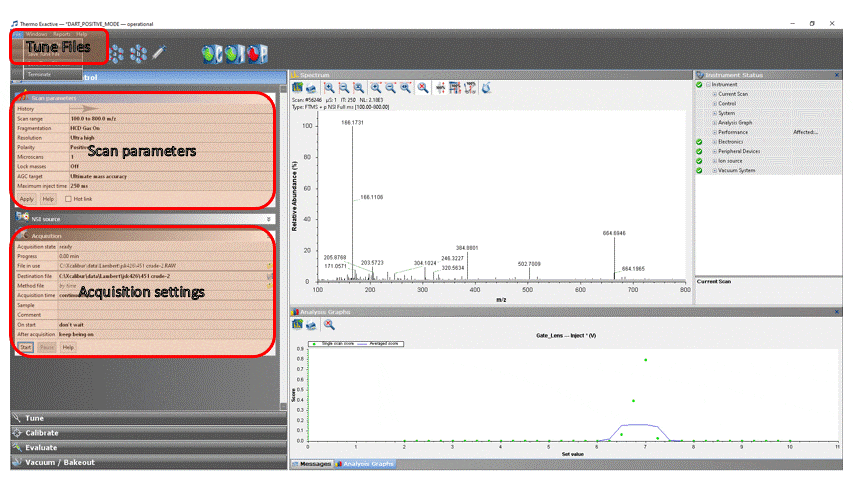
- Load the appropriate DART method into ExactiveTune by clicking File → Load Tune File… → DART_POSITIVE_MODE for positive ion analysis or DART_NEGATIVE_MODE for negative ion analysis.
- Modify the scan parameters if desired, but do NOT change ANY settings not specifically listed below without permission from facility staff![1]
To adjust a parameter, click on the value, change it to the desired setting, then click the “Apply” button.
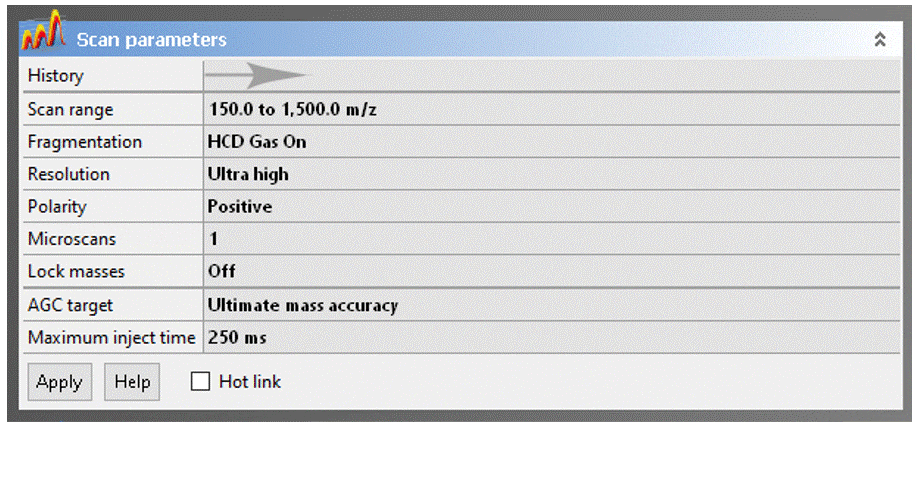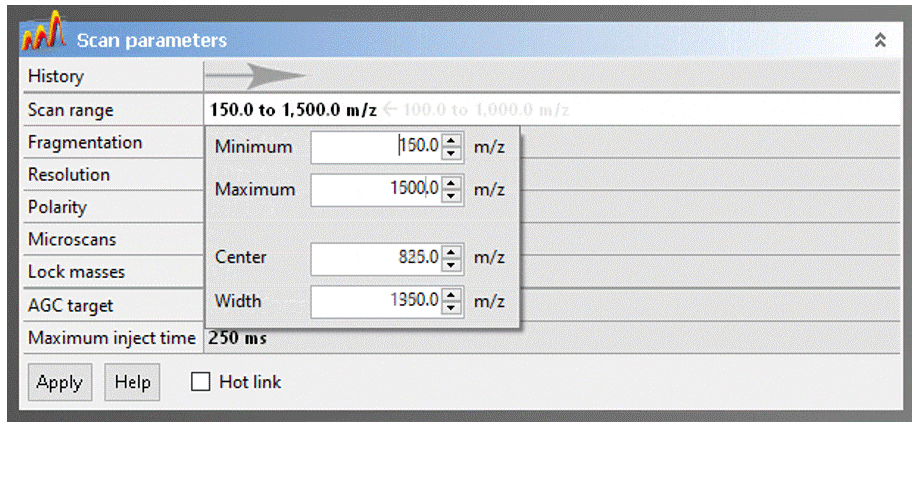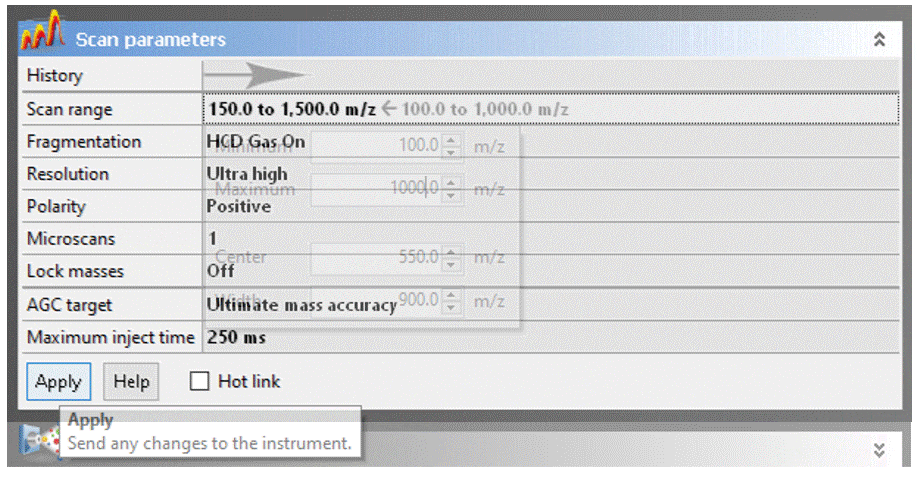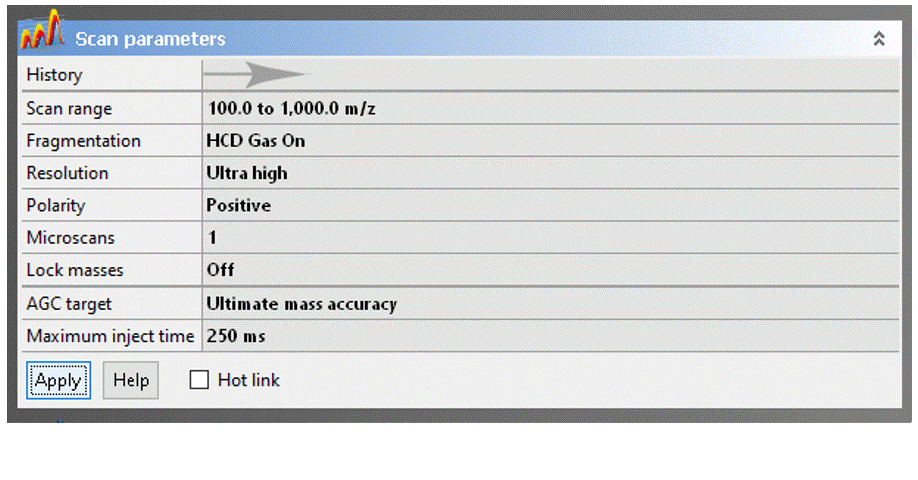
- Scan range: The Exactive has a calibrated m/z range of 50–2000. You may adjust to any desired window keeping in mind that:
- Reducing the the scan range has no effect on resolution or mass accuracy.
- If you have intense low-mass ions (m/z < ~150), setting the scan range to start a few Daltons above their mass may give you better sensitivity for higher masses.
- Eliminating higher masses has no effect on sensitivity
- DART will typically not ionize compounds with MW much greater than ~1000. Exceptions to this rule are unusually volatile molecules, like organometallic coordination complexes and polyalkylsiloxylated molecules.
- Resolution: You can sacrifice resolution to increase scan rate, which is never a good trade off for DART analyses. This setting is provided for UHPLC and nanoLC ESI applications, where higher scan rates are necessary for accurate detection of chromatographic peaks.
- Polarity: Specifies positive or negative ion MS, but you should not change this field! To switch between ion modes, load the appropriate Tune file (see above).
- Gain: Change ONLY if higher dynamic range is desired to improve sensitivity for trace analytes in the presence of much stronger signals.
- Scan range: The Exactive has a calibrated m/z range of 50–2000. You may adjust to any desired window keeping in mind that:
- Press the Apply button to upload your changes to the Exactive. Nothing changes until you click “apply.”
- In the ExactiveTune software, select the Acquisition tab. Edit the parameters as follows (ones in italics are unchanged and are only listed here for your reference):
Acquisition → ready
File in use → The path and filename of the last destination file is displayed here
Destination file → Click the “floppy disk” icon and navigate to “This PC” → OS (C:) → Xcalibur → your group → your NetID and enter a file name. You can create the directories if they don’t exist. NOTE: You cannot save the data directly to the fileshare (drive W:), you will need to copy the file manually when you are done.
Method file → by time
Sample → Optional information about your sample (notebook number, reference, lot #, etc.)
Comment → Optional comments about your sample (any text you’d like)
On start → don’t wait
After acquisition → 10 minutes (you can always stop it earlier, but this ensures that if you forget to click stop your experiment won’t fill the hard disk) - Get the mass of the plasticizer!
The capillary is coated with dioctyl adipate, a common plasticizer is soft plastics. We strongly recommend that you record its mass so you can use it as a reference in your analysis.- Turn the DART gas temperature to 150-200°C
- Click the “Start” button in the “Acquisition” tab in Exactive tune. At this point, the MS will save the collected MS data until you click the “Stop” button.
- Take a fresh capillary from the container. Be careful not to touch the closed end. Hold the capillary between the white insulators and look for a m/z around 371.3156. (Disregard the peak at 371.1, that’s something else.) If you can’t see the plasticizer, please come and get us. You may need to lightly rest your hand on the DART to keep the capillary steady. If the plasticizer is outside the 371.3126 to 371.3186 range, please let us know so we can recalibrate the spectrometer.
- Acquire the data for your sample and save the file.
- Adjust the DART gas temperature to 50-100°C below the molecular weight of your analyte, or 200°C, whichever is lower.
- Sample your analyte with the same capillary that you used to record the adipate. If you are using a solution allow the capillary to briefly air dry.
- If your sample is a solid, pick up a very small amount with the closed end of the capillary. If you can obviously see solid on the tip, you picked up too much! Be sure there are no solid “chunks” of sample present. These will contaminate the source with your sample for quite some time.
- Check for the presence of analyte ions by waving the capillary through the DART source. If waving doesn’t produce very strong signals hold the capillary in the sampling region. You can move the capillary around the sampling region but please be very careful NOT to touch the white insulators. You may need to lightly rest your hand on the DART to keep the capillary steady. Slowly turning the capillary between your fingers can also be helpful.
- Repeat the process two or three times. This will make it easier to differentiate analytes from background and contamination during processing.
- OFF, STOP, PAY!
- Click “Off” in the DART window
- Click “Stop” in the Exactive software
- Pay for the samples you ran at the laptop by the Orbi
- Mac users ONLY:
Open your data in MNova, make sure the “Fetch Full Dataset” has a blue arrow pointing up, and save the MNova file. - Transfer your data to the NMR fileshare using File Explorer. You can create a new subfolder for your MS data in your data folder (W:\Group\NetID), but you won’t be able to rename it from the mass spec computer. You can rename the folder later from your own computer when you download the data. Process your data using the MNova MSChrom plugin that is included in the MNova download by default. You can download the license here.
Reporting DART data
You can describe the instrument in experimental sections of publications as “Mass spectra were acquired on a DART-SVP (Direct Analysis in Real Time) ion source (IonSense, Saugus, MA) coupled to an Exactive Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany).” Report HRMS data as “HRMS (DART/Orbitrap) m/z: [M + H]+ Calcd for C22H43O4 371.3156; Found 371.3155.” Note that journals have differing requirements on the accuracy of HRMS data. The Journal of Organic Chemistry, for example, suggests an error of less than 3 mmu, which should be easily achievable. Organic Letters still demands less than 5 ppm, which can be tricky for low molecular weight compounds. JACS doesn’t appear to have any specific requirements. Always check the instructions for authors before submitting to a publication.
[1]Experience teaches us that if it doesn’t work, clicking around randomly will not fix it. It will make it worse. We have seen this time and time again, so please trust us on this.